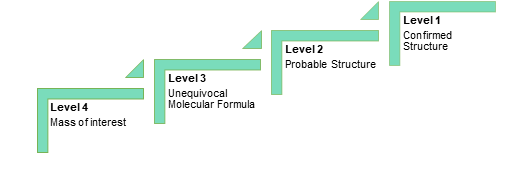
Qualimetrix is a customer-driven Testing Laboratory that employs a structured and well-defined approach in order to design and implement optimized processes with the aim of transforming customer inputs and requirements into “customer value”. As such, the first and probably the most critical factor for a successful project is its proper definition in terms of both customer and technical requirements. To this end, a comprehensive study request form is provided to the customer with the following objectives:
- The definition of the type and scope of the study
- The provision of critical product information
- The determination of the most suitable, expedient and cost-effective approach
The following figure presents a project setup; meaning the logical process by which a series of studies may be proposed to a sponsor.
Polymeric and elastomeric materials are commonly encountered during the manufacturing process of pharmaceutical products as well as components of the packaging / container closure system. During the product’s expected shelf-life and use, the constant contact, as well as the stressing, may bring about a change in the composition of the product stored, through interactions with its packaging.
Product packaging interaction studies focus on establishing this change in product composition brought about by the interplay of packaging and stored content through means of molecular exchange. This exchange involves the solubilization of compounds within the polymeric or metal matrix and their subsequent migration into the bulk of the stored product.
The main phases of interest in the product's life cycle that are relevant to drug-packaging interaction studies and hence to the safety assessment are schematically presented in the figure below and explained in more detail in the following paragraphs:
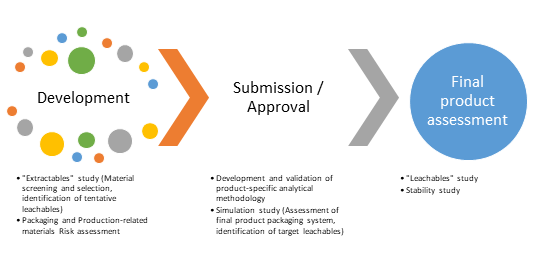
- Development: The first operational step related to product-packaging interaction studies is the material screening process. During this process, all candidate materials are evaluated in terms of their available information. The means by which this initial evaluation is performed are risk assessment and gap analysis, compendial testing, which is usually conducted by the supplier and extraction studies in order to establish the material composition and mitigate any information / data gaps.
“Extractables” study
Controlled Extraction studies are of paramount importance in order to:
- Characterize candidate materials and assess their suitability for use
- Cover the safety gaps resulting from the lack of compendial testing or other material information that, in many cases, the suppliers are not eager to provide.
- Identify “tentative” leachables that could be employed as target analytes for the development and validation of a “product-specific” methodology for the determination of leachables
The applied semi-quantitative generic methodology has been designed to cover representative leachables, designated by extraction studies of packaging materials available and which are commonly used in plastic manufacturing. The purpose of the initial screening of extractables is mainly to establish a “worst-case” potential leachables profile for the product-specific packaging materials and facilitate the establishment of qualitative and quantitative leachable-extractables correlations.
Extraction techniques commonly employed for this initial step include but are not limited to the following:
- Maceration (solvent soaking)
- Reflux
- Soxhlet
- Sonication
- Sealed vessel
The profile of the extractable components is acquired by the use of leading edge, hyphenated, orthogonal analytical techniques, required to cover their significant chemical diversity (e.g. LC-HRMS, GC/MS, GC/FID, ICP/MS etc.). Extractions that are not solvent-mediated can also be performed through the use of Headspace Gas Chromatography (HS-GC/MS).
Packaging and Production-related materials risk assessment
A preliminary assessment of the extent of component testing is necessary in order to establish the suitability of plastic components involved in both packaging and the manufacturing process stream (e.g. tubings, filters, connectors, etc.). The assessment is based on risk factors related to the nature and conditions of the contact between the product stream, the extraction propensity of the solvents used and the nature of the plastic materials.
Recently, a framework for the risk assessment conducted for leachables in pharmaceuticals has been introduced. The Extractables and Leachables Safety Information Exchange (ELSIE) group has proposed such a framework, based on the concepts of the ICH Q9 Quality Risk Management guideline, while the general chapter of USP–NF 2022, Issue 1, <1665> proposes a framework for the risk evaluation of production related materials prior to the design of studies that address this risk or gaps in data that hinder the risk evaluation process.
An evaluation strategy and a risk rubric proposed in relevant scientific literature has been reviewed, evaluated and properly amended by our scientific team, in order to form the “backbone” of risk assessment for a pharmaceutical product’s leachable species profile.
The actions that need to be taken for risk mitigation / reduction and acceptance are presented within the concluding section of the risk assessment report.
The Risk Assessment exercise for Leachable Species in Pharmaceutical Products” is a very demanding process considering that it is not restricted to data presentation but also inference based on existing data. Depending on the reliability of the existing data and/ or constraints regarding the conditions/ processes/ materials that allow safeguarding quality, the risk assessment process may limit or even waive the required testing. For example, a low maximum dose pharmaceutical product, with a composition that does not exhibit a high propensity for leachable species solubilization, is likely to arrive to a “leachable species profiling” waiver through the risk assessment process – making the risk assessment procedure very cost-efficient. A high dose liquid injectable product on the other hand may even require testing through its shelf-life for determining the kinetics of migration and modifying the shelf-life appropriately so as to mitigate the risk of critical exposure.
- Submission and Approval: This phase reflects a product that is fully defined and completely characterized with respect to leachables. This practically means that a leachables study has already been performed on the final product by employing a validated method in order to establish the product's leachables profile.
Development and validation of product-specific analytical methodology
Based on the results obtained from the extraction study (or simulation study) previously performed, the generic methodology, comprised of the sample pre-treatment and analysis stages, is properly adjusted in order to become a "tailor-made" product-specific methodology targeting the analytes / potential leachable species, identified during the extraction study that exceed or have the potential to exceed the product's Analytical Evaluation Threshold (AET) during the actual leachables study. The next step is to make this "tailor-made" method also "fit for purpose" by means of method validation according to the principles set by ICH Q2 (R1) guideline.
The validated product-specific methodology is subsequently applied in order to perform the actual "leachables" testing and provide reliable quantitative results for the leachables of interest.
Simulation study (Assessment of final product packaging system, identification of target leachables)
However, since the leachables assessment should cover the product's shelf-life, it is rather hard to have relevant data available at the time of submission. To this end, simulation studies can be performed as a "surrogate" by submitting the final product to elevated temperature conditions in order to simulate the anticipated stressing effect at the end of shelf-life.
Moreover, simulation studies where the actual drug product is replaced by a solvent of equal or similar propensity can be performed in the following cases:
- Drug products with an extremely complex and challenging matrix (e.g. lipid emulsions) where a more “analytically expedient” sample needs to be produced for the evaluation of “leachables”
- Identification of “probable” leachables that could be employed as target analytes for the development and validation of a “product-specific” methodology for the determination of leachables. The advantage versus the extraction study is that the long list of “extractables” is significantly reduced and the target analytes are much more relevant since the simulation study mimics the conditions experienced by the final drug product
- Final Product Assessment and Maintenance: This phase mainly comprises of the final and definitive assessment of the product at the actual end of shelf-life as well as issues that may arise from vendor-related raw material or compositional changes that may have an impact on the leachable species profile (change control)
"Leachables" study
Application of the validated “product-specific” analytical methods for the quantification of leachables in the final drug product, stored under normal and accelerated storage conditions (e.g. ICH conditions) at the end of the product’s shelf-life. Both target analytes, previously identified from extraction / simulation studies, and secondary leachables are monitored and determined.
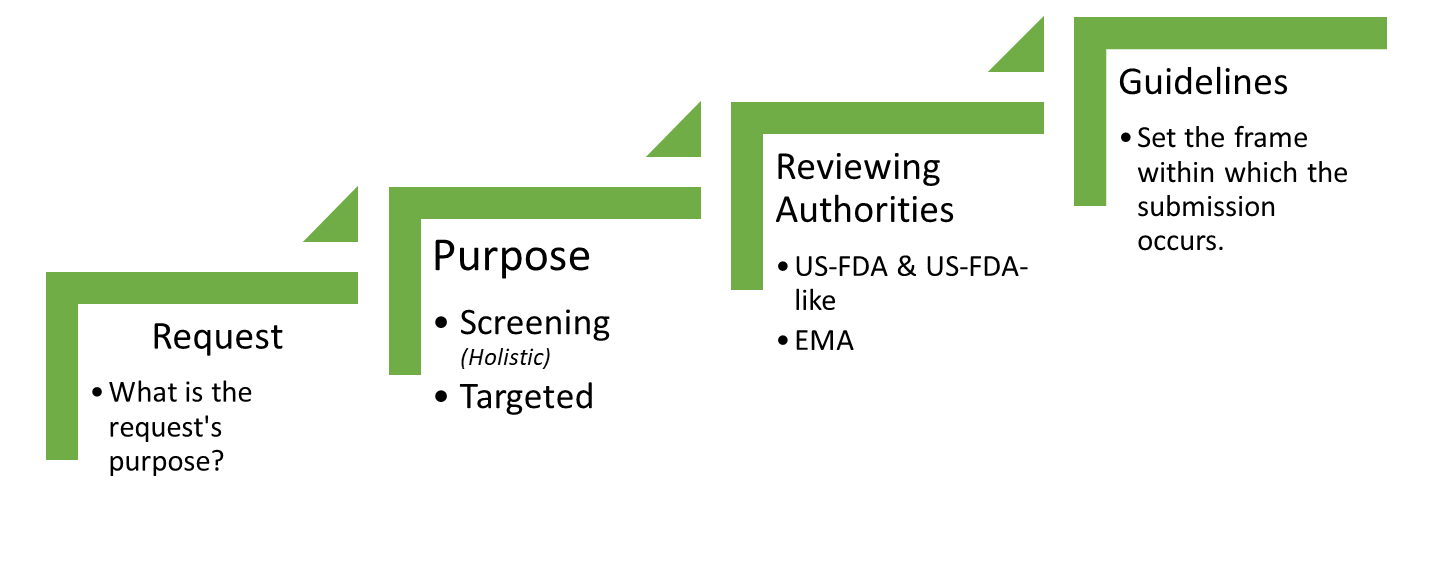
On occasion, the authorities may express a request that is actually targeted on a specific substance or a group of substances. Based on the request, this may fall either under screening studies or targeted studies.
Typical examples include the following:
- OVIs – Organic Volatile Impurities: This is usually triggered by the use of a rubber material in the product. It is addressed through a screening study but, as implied from the nomenclature, it is limited to volatile organic species.
- Bis(2-ethylhexyl) phthalate (DEHP): A known endocrine disruptor for which a high exposure of the general population is suspected. The request is usually triggered by the use of PVC materials in either manufacture, storage or even dilution of a pharmaceutical product. This is a targeted request.
- Benzophenone: A known potent photosensitizer species. The request is triggered by the submission of data on labeling to the authorities, that implies or states the use of a benzophenone or acetophenone photoinitiator. Depending on whether the data declare the exact compound used or not, the study may be treated as a targeted study or a screening study.
The techniques employed include:
- LC-PDA- (ESI/ APCI) HRMS: A combination of liquid chromatography with mass spectrometry and UV/ vis-based detection. Contrary to conventional LC-UV chromatography, detection is not highly strained by co-elution due to HRMS’s capability for high resolution based on ion m/z. Species detectable correspond primarily to low volatility analytes with chemical moieties that are amenable to ionization or dipole induction; this practically includes almost all of the potentially bio-active chemical species: amines, amides, esters, alcohols, acids, ketones, sulfates/ sulfites, phosphates/ phosphites, etc.
- (HS/L) GC-(EI/CI) MS: A combination of gas chromatography with mass spectrometry that allows sample introduction either in gas form (upon thermal desorption) or liquid form. The technique may employ electron impact or chemical ionization, with an emphasis on EI due to the extensive fragmentation libraries that exist under the convention of using a 70 eV field. Detectable species range from highly to semi-volatile species and while partial overlap with LC-HRMS is possible when targeting the “semi-volatile” range, GC-MS is necessary to allow the detection of non-polarizable species i.e. hydrocarbons, polymer oligomers, ethers, sulfides, halogenated hydrocarbons, etc.
- ICP-MS: The successor to classic FAAS techniques that allows targeting of multiple elements in a single analytical run without the need for element-specific lamps. The detection of elemental impurities is based on the characteristic m/z values for their isotopes, while interference and instability due to sample composition is minimized due to the atomization process and the collision / reaction cell technology employed.
Evaluation of the methods based on the above, or other techniques if required (ion chromatography, size-exclusion chromatography, etc.), is mainly based on the principles of ICH Q2 (R1) guideline.